 Reviewed by Mr. Ranjeet Kumar
Sr. Audiologist, Speech Therapist & Cochlear Implant Specialist, BASLP on
Reviewed by Mr. Ranjeet Kumar
Sr. Audiologist, Speech Therapist & Cochlear Implant Specialist, BASLP on
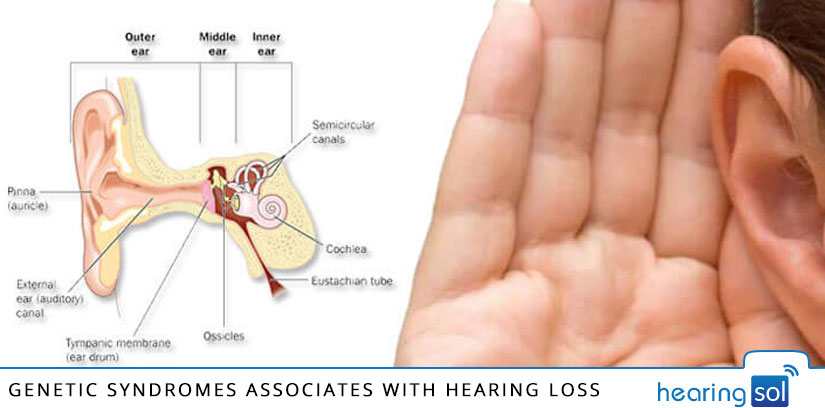
Hearing Loss, the word itself defines the condition. Once the condition is noticed, consulting an audiologist or hearing specialist is the best option.
But, what if an audiologist is unable to determine the cause of your hearing loss? What if some other signs and symptoms are also noticeable along with the hearing defect?
You can purchase the latest hearing aids at a fair price through HearingSol, If you need any assistance or you have a query regarding Genetic Syndromes and Hearing Loss, feel free to call us at +91-9327901950. We are always here to help you.
10 Genetic Syndromes
To find out the answers to the following question, you are advised to consult a geneticist. Because your hearing defect may be the outcome of a genetic condition/syndrome.
As there are many genetic syndromes associated with Hearing Loss, a geneticist will be the best person to consult.
1. Alport Syndrome
Alport syndrome is defined as a genetic condition which is characterized by the following effects kidney disease, hearing loss, as well as eye abnormalities.
People having this syndrome experience progressive loss of kidney function. Almost all the people affected with syndrome experience blood in their urine i.e. hematuria. This condition indicates abnormal kidney functioning.
Along with kidney, eyes and hearing are also affected by this syndrome in many patients. October is celebrated as the month of Hearing Loss Awareness which is the perfect time to check the effect of Alport syndrome on your hearing.
Alport syndrome can also be responsible for causing hearing loss although some patients may not be affected.
People having Alport syndrome are born with normal hearing, hearing loss develops progressively, usually having normal kidney functioning at that stage. But there may be substantial proteinuria.
In some conditions, hearing loss is noticed after the loss of kidney functioning. The early changes in this condition have reduced the ability to hear high-frequency sounds leading to ‘high-tone hearing loss’.
Later, this condition becomes more severe and affects lower frequencies too. In Alport syndrome, hearing loss is not complete. It can be overcome with the use of hearing aids.
In people with Alport syndrome, the collagen makeup of the basement membranes of the cochleas or inner ears is abnormal. However, the cause of hearing loss is still not known, or whether it can be prevented or reversed.
Among all the types of hearing loss, the Bilateral sensorineural hearing loss is among the most initial symptom to occur.
A person having this type of hearing loss will face difficulty in hearing and understanding speech or interpreting sounds, and especially over background noise or when the speaker mumbles.
2. Branchio-Oto-Renal Syndrome
Branchio-oto-renal syndrome is also known as the BOR syndrome. This syndrome is an autosomal dominant genetic disorder which affects kidneys, ears, and neck. It is also described as Melnick-Fraser syndrome.
Ear anomalies such as extra openings in the front portion of the ears, extra growth pieces of skin in front of the ears, and even malformation or absence of the outer ear known as the pinna.
Due to the presence of this syndrome, malformation, or absence of the middle ear part is also possible. The individuals suffering from Branchio-oto-renal syndrome (BOR) can have mild to profound hearing loss.
Signs and symptoms of branchiootorenal syndrome or BOR can vary from person to person. Symptoms can vary even between individuals belonging to the same family.
Hearing loss is among the most common symptom witnessed in BOR syndrome and can be noticed in approximately 90% of people with this syndrome.
The type of hearing loss may differ based on the condition i.e. conductive, sensorineural, or a combination of both (in some cases).
Other signs and symptoms of this syndrome are branchial cleft cysts, branchial fistula, outer, middle or inner ear malformations, and kidney malformations.
Mutations of genes, such as EYA1, SIX1, and SIX5, are the known cause of the branchiootorenal syndrome.
Hereditary hearing loss conditions tend to be managed by a team including an otolaryngologist, an audiologist, a clinical geneticist, a neurologist, and a deaf educator in some conditions.
People having hereditary hearing loss require regular follow-up with a hearing specialist for monitoring stability or progression of the hearing loss.
This type of hearing loss can be treated with the help of hearing aids or vibrotactile devices. In some cases, cochlear implantation can also be considered for children over the age of 12 months having a severe-to-profound hearing loss.
Early hearing intervention in such cases is recommended, especially for children who are at risk of losing their hearing before they learn to speak. This can be done through amplification, surgery, or cochlear implantation.
3. CHARGE Syndrome
CHARGE syndrome is a type of disorder which affect many areas of the human body. This syndrome was earlier known as CHARGE association, which indicated a certain pattern of congenital anomalies/defects which occurs together more frequently than expected on the basis of chance.
CHARGE is an abbreviation created in 1981 by R. A. Pagon and her coworkers, used for several common features witnessed in this disorder:
- C – Coloboma of the eye, central nervous system anomalies
- H – Heart defects
- A – Atresia choanae (also known as choanal atresia)
- R – Retardation of growth or development
- G – Genital defects such as Hypogonadism, undescended testicles, besides hypospadias
- E – Ear anomalies or deafness and also bowl-shaped and concave ears which are known as “lop ears”.
It is a rear syndrome caused due to some genetic disorder and occurs only in 0.1–1.2 per 10,000 live births. As of the year 2009, CHARGE syndrome was the leading cause of congenital deafblindness in the US population.
The acronym “CHARGE” was first used in 1979, for a newborn child having the following congenital features of Coloboma of the eye, Heart defects, Atresia of the nasal choanae, Retardation of growth or development, Genital abnormalities, and Ear abnormalities or deafness.
The following features are no longer in use for diagnosing the CHARGE syndrome, but the name remains.
Almost 2/3rd cases of this syndrome are due to mutation of the CHD7 gene (located on chromosome 8). Complete deafness and blindness are rare in this syndrome.
The most common feature witnessed in this syndrome is a partial loss of hearing as well as vision. Almost half of the individuals suffering from CHARGE syndrome have a severe-to-profound hearing loss.
Many of these individuals with hearing anomalies are really difficult to evaluate as suggested by audiologists.
In CHARGE syndrome almost every set of auditory system structures may be involved. External ears are malformed and have malformation patterns which are so distinctive that preemptive diagnosis of CHARGE is done on the basis of unusually shaped, asymmetrical pinnae.
Middle ears typically showcase ossicular malformations which results in the conductive hearing loss that is greatest in the low frequencies but is present at every frequency.
4. Crouzon Syndrome
Crouzon syndrome is described as a genetic disorder which is characterized by the premature fusion of certain bones present in the skull. This condition is also known as craniosynostosis. Normal growth of skull is prevented and shape of the head and face is affected, due to this early fusion.
This premature fusion of skull bones results in many other features of this syndrome.
The abnormal growth and premature fusion of the bones lead to many other conditions such as wide-set, bulging eyes and vision problems i.e. caused by shallow eye sockets; a beaked nose; and an underdeveloped upper jaw.
Due to the shallow eye socket, eyes do not point in the same direction, and this condition is known as strabismus. In addition, people with Crouzon syndrome may have dental problems and hearing loss, which is also sometimes accompanied by narrow ear canals.
Patients suffering from Crouzon syndrome may exhibit malformations of the external and/or the middle ear, such as malalignment of the pinna.
They typically suffer from the conductive hearing loss which is caused by middle ear effusion (or fluid in the middle ear), perforation to ossicular fixation, tympanic membrane, ossicular anomalies, and also the closure of the oval window.
Patients suffering from sensorineural hearing loss are less likely to occur but still, have been reported.
Some people suffering from Crouzon syndrome have an opening in the lip as well as the roof of the mouth. This is known as cleft lip and palate.
The severity of the signs and symptoms of this syndrome vary from person to person. The person having Crouzon syndrome, usually are of normal intelligence.
The condition is inherited in an autosomal dominant pattern. This means that a single copy of the altered/mutated gene in each cell is sufficient to cause the disorder.
There are two genes associated with Crouzon syndrome i.e. FGFR2 and FGFR3. FGFR is used for Fibroblast Growth Factor Receptor.
There are many fibroblast growth factor receptor genes which are numbered 1 to 4. The following genes are also involved in bone growth. The most common gene associated with Crouzon syndrome is the FGFR2 gene.
5. Down’s Syndrome
Down’s syndrome is described as a genetic disorder. This disorder is caused when there is an abnormal cell division which results in extra genetic material from the chromosome 21.
This syndrome causes a distinct facial appearance, intellectual disability, and even developmental delays for those with Down’s Syndrome. It may be sometimes associated with thyroid or heart disease.
People with Down’s Syndrome have a significantly higher chance of suffering from hearing problems as compared to other groups. The hearing problem may be a mild temporary hearing loss or a longer-term problem.
The main cause of the hearing defect is a build-up of fluid in the space behind the eardrum (i.e. glue ear, also referred to as Otitis Media with Effusion OME). This can also be due to a build-up of ear wax or an ear infection.
The importance of hearing cannot be overemphasized. As suggested by some survey, almost 80% of people with Down syndrome will have some hearing problem.
For a child with Down syndrome, early detection, and treatment of hearing deficits with a team of therapists and special educators is really essential. This can help the sufferer to manage the Down’s syndrome.
Here are some of the causes for higher levels of hearing the loss in people with Down’s syndrome:
- increased incidence of chronic ear diseases
- differences in the structure of the ear
- weaker immune systems.
6. Goldenhar Syndrome
Goldenhar Syndrome is also known as hemifacial microsomia and oculoauriculovertebral spectrum or OAV spectrum/displaysia. Most of the sufferers have a malformed outer ear, a condition known as microtia. In almost one-third of the cases, microtia is bilateral and is rated on a four-point scale.
These malformations can be present in any form such as preauricular ear pits, the absence of the auricle, stenosis or atresia of the external auditory canal, ossicular malformations, middle ear deformities, and incomplete pneumatization of the temporal bone. Almost 95% of the sufferers have some type of ear malformation.
Additionally, a conductive hearing loss can also be present which can range from mild to severe. Almost 86% of the patients have a conductive hearing loss due to malformed middle ear bones and 10% have a sensorineural hearing loss, even cases of cochlear involvement are also reported.
As there is no correlation between the severity of dysmorphic features and the degree of hearing loss, which means that individuals with mild malformations may have severely impaired hearing.
Goldenhar is a group of conditions which is known as craniofacial conditions. That means the impact of the syndrome is primarily on the head and face.
In almost 85% of patients, only one side of the face is affected i.e. one side of the face looks smaller as compared to the other.
In some cases, there may be an incomplete development of the facial muscles, cleft lip or palate (which may appear like an abnormally wide mouth), and teeth abnormalities.
The possibility of vertebral malformations can be there in Goldenhar Syndrome. In this case, the vertebrae of the sufferer may be underdeveloped, fused, or missing.
Some children with Goldenhar Syndrome may have heart, lung, kidney or gastrointestinal problems, and even genital malformation. There is a certain percentage of sufferers who may have mental retardation.
7. Pendred Syndrome
Pendred syndrome is described as a genetic disorder which can lead to congenital bilateral sensorineural hearing loss and goiter with euthyroid or mild hypothyroidism (i.e. decreased thyroid gland function).
There is no specific treatment for this syndrome, other than supportive measures used for the hearing loss and thyroid hormone supplements in case of hypothyroidism.
This syndrome is named after Dr. Vaughan Pendred (1869–1946), an English doctor who first described this condition to an Irish family from Durham in the year 1896.
This syndrome accounts for 7.5% to 15% of all cases of congenital deafness reported. The hearing loss of Pendred syndrome is not always present from birth.
But language acquisition may be a significant issue if the hearing disorder is severe in childhood. This hearing defect typically worsens over the years.
The progression can be stepwise and related to minor head trauma. There are some cases where language development worsens after a head injury.
This demonstrates that the inner ear is sensitive to trauma in the case of Pendred syndrome. This is a consequence of the widened vestibular aqueducts which is usual in this syndrome.
In the case of Pendred syndrome, Vestibular function varies and vertigo can be a feature of minor head trauma. The condition of goiter is reported in almost 75% of all cases.
Usually, the audiometry is abnormal, but the findings are still not very specific. Therefore, an audiogram is not sufficient for the diagnosis of this syndrome.
8. Stickler Syndrome
In case of Stickler syndrome, the sensorineural hearing loss is the most typical hearing loss witnessed in patients, which indicates that the source of the deficit lies in the inner ear, the vestibulocochlear nerve or the processing centers of the brain.
In the year 2001, Szymko-Bennett et al. found out that generally the overall hearing loss in Type I Stickler Syndrome is mild and is not significantly progressive. In the higher frequencies i.e. from 4000–8000 Hz, hearing loss is more common.
This syndrome is thought to be caused due to mutation of several collagen genes while fetal development is going on. This trait is sex independent autosomal dominant.
This means that a person with this syndrome has about a 50% chance of passing it on to their every child. Stickler syndrome is caused by the mutation in the COL11A1, COL11A2, and COL2A1 genes. Type II and type XI collagen are produced with the involvement of these genes.
About 80% of patients suffering from Stickler syndrome, mildly progressive sensorineural loss, or more significant losses may be present which is associated with Types II and III Stickler syndrome.
However, there are other patients who are susceptible to conductive losses, which is also similar to nonsyndromic cleft patients. Children having a cleft palate are prone to ear infections and also swallowing difficulties in some conditions.
9. Treacher Collins Syndrome
It can be also referred to as TCS or Franceschetti-Zwahlen-Klein syndrome or mandibulofacial dysostosis. It is named after Edward Treacher Collins, who was an English surgeon, and an ophthalmologist as he described this syndrome in the year 1900.
This syndrome occurs in approximately 1 in every 50,000 births. People suffering from Treacher Collins syndrome often showcase both cleft palate as well as hearing loss, in addition to other disabilities.
In this syndrome, Hearing loss is either secondary or absent, small or unusually formed ears (i.e. microtia) which commonly results from malformations of the middle ear.
According to the research, most of the patients suffering from Treacher Collins syndrome have symmetric external ear canal abnormalities i.e. symmetrically dysmorphic or sometimes absent ossicles in the middle ear space.
In most of the cases, Inner ear structure is normal. Most patients show a moderate or greater hearing impairment, and generally, there is a conductive hearing loss.
People with Treacher Collins syndrome showcase hearing losses which is similar to those of patients with malformed, abnormal, or missing ossicles.
This syndrome is caused due to the mutation of the TCOF1 gene. The mutation may be inherited by the children from their parents, or it may be a spontaneous occurrence.
It is an autosomal dominant disorder. There are 50% chances that an individual having the TCOF1 mutation will pass it to their children. This is the reason for Treacher Collins syndrome being hereditary.
10. Waardenburg Syndrome
Waardenburg syndrome is a group of genetic conditions that was first described in the year 1951. These genetic conditions can cause varying degrees of hearing loss, minor structural defects arising from the neural crest, changes in (pigmentation) hair color, skin color, and eye color.
Waardenburg syndrome is caused due to Mutations in the EDN3, EDNRB, MITF, PAX3, SNAI2, and SOX10 genes. These genes are involved in the formation and development of melanin/pigment-producing cells i.e. Melanocytes.
It also plays an important role in the normal functioning of the inner ear as well as the development of nerve cells in the large intestine.
The mutation disrupts normal hearing and normal development of melanocytes which leads to abnormal pigmentation of the skin, hair, and eyes.
Mostly, people with Waardenburg syndrome have normal hearing but can have a moderate to a profound hearing loss in one or both ears. The hearing loss is congenital (i.e. present from birth).
People with this syndrome have pale blue eyes or different colored eyes i.e. one blue and one brown eye. In some conditions, one eye can have two different colors in segments, and the same with the hair color.
In Waardenburg syndrome features vary from person to person, even if they belong to the same family.
Later this syndrome was found to have four types based on their physical characteristics and their genetic cause.
- Type I – Though, type I and II have similar features, but people with type I always possess eyes that appear widely spaced. In this type, Hearing loss is less often. This type is caused by mutations in the PAX3 gene.
- Type II – It was first identified in the year 1971. Hearing loss is more common in this type. Widely spaced eyes are not witnessed in type II Waardenburg syndrome. This type is caused by mutations in the MITF or SNAI2 gene. In some cases, Type II along with type IV appears to have an autosomal recessive pattern of inheritance i.e. both copies of the gene in each cell are mutated.
- Type III – It is also known as Klein- Waardenburg syndrome. This includes abnormalities of arms and hands along with hearing loss and pigmentation change. This type is caused by mutations in the PAX3 gene.
- Type IV – It is also known as Waardenburg-Shah syndrome. It has signs and symptoms of both Waardenburg syndrome and Hirschsprung disease. Hirschsprung disease is an intestinal disorder that causes severe constipation or intestine blockage. This type is caused by mutations in the SOX10, EDN3, or EDNRB gene.
All the syndromes or genetic syndromes described above are related to hearing loss/hearing disorder in some, or the other way. These syndromes may play a contributing factor to the hearing defect, or vice-versa.
Apart from the above-mentioned syndromes, there are some more added to the list: Jervell and Lange Nielsen syndrome, Usher syndrome, Norrie disease, Perrault syndrome, Cornelia de Lange Syndrome, Fetal Alcohol Syndrome, Pierre Robin Sequence, Marshall Syndrome, Rubella Syndrome, etc.
You can purchase the latest hearing aids at a fair price through HearingSol, If you need any assistance or you have a query regarding Genetic Syndromes and Hearing Loss, feel free to call us at +91-9327901950. We are always here to help you.
Read Also: